
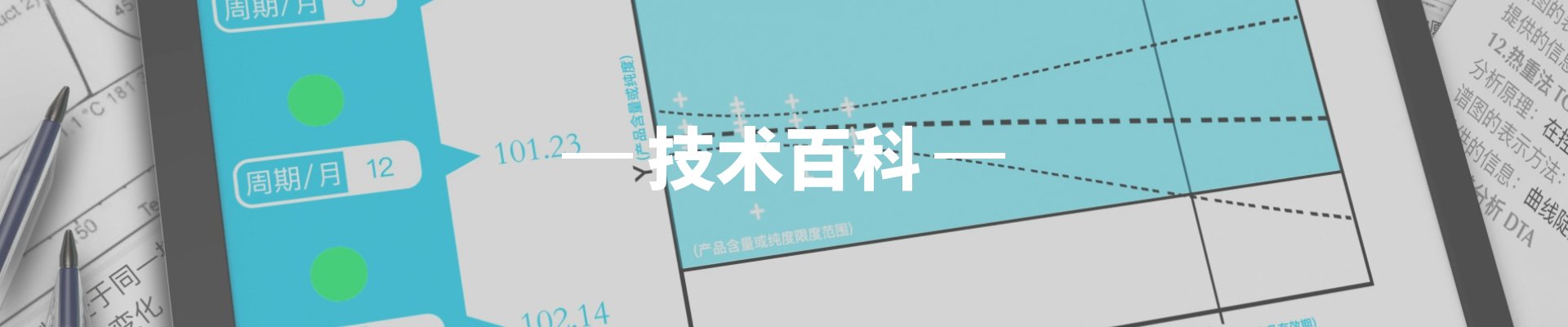
杂质控制限度是药品质量控制中的重要参数,对保证药品安全性和有效性具有重要意义。杂质控制不仅涉及原料药的质量标准,还包括制剂过程中可能产生的降解产物和相关物质的控制。
在药品研发和生产过程中,杂质的来源主要包括以下几个方面:合成过程中的副产物、起始原料及中间体残留、制备过程中使用的试剂和溶剂残留、储存过程中的降解产物等。这些杂质可能会影响药品的安全性和有效性,因此必须严格控制其含量。
杂质控制限度的制定需要考虑多个因素。首先是毒理学评价,需要根据杂质的结构特征、致突变性、致畸性等毒理学数据来确定其安全限值。其次是分析方法学考察,采用的检测方法必须具有足够的灵敏度和选择性,能够准确定量目标杂质。此外,还要考虑生产工艺的能力和稳定性,确保在常规生产中能够持续达到规定的杂质控制要求。
国际人用药品注册技术协调会(ICH)对药品杂质控制提出了详细的指导原则。根据ICH Q3A和Q3B指导原则,对于日剂量大于2g的药物,已知杂质的控制限度通常不得超过0.05%;对于日剂量小于2g的药物,需要根据最大日剂量计算杂质的允许摄入量。对于未知杂质,其控制限度通常应更严格,一般要求不得超过0.1%。
在实际生产中,杂质控制需要建立完善的质量控制体系。这包括原料进厚控制、工艺过程控制和成品检验等多个环节。需要通过验证性试验确认工艺的稳定性和可靠性,同时建立适当的检测方法来监控各类杂质的含量。对于重要的杂质,可能需要采用先进的分析技术,如高效液相色谱、气相色谱、质谱等方法进行定性定量分析。
此外,药品在储存过程中可能会产生降解杂质,因此还需要通过稳定性研究来考察储存条件对杂质谱的影响。这要求在产品的有效期内定期检测杂质含量的变化,确保其始终符合质量标准的要求。如果发现某些杂质含量有增长趋势,则需要及时采取措施,如改进包装材料、优化储存条件等。
随着分析技术的进步和法规要求的提高,杂质控制限度的制定和执行将更加严格和科学。这需要制药企业不断提升研发和生产能力,确保药品质量始终符合相关法规要求,为患者用药安全提供可靠保障。

上一篇:工艺杂质和降解杂质的区别
下一篇:没有了
Copyright © 2023-现在 广州佳途科技股份有限公司 版权所有 粤ICP备18108372号-4
CATO标准品信息网为您提供全面标准品、对照品、药物杂质等产品信息,包括品牌、规格、报价等信息,满足实验中标准品/药物杂质采购需求。
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F 电话:020-81960175-877
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F
电话:020-81960175-877
企业邮箱:pinfangshu@cato-chem.com 业务QQ:2853283383
业务QQ:2853283383
![[52317-98-3] N-甲基-3-戊胺](https://www.cato-chem.com/files/3,2b23ff66c8d0e8.jpg)
![[1772607-47-2] 尿苷二磷酸胆碱(UDPC)钠盐](https://www.cato-chem.com/files/4,2b2d4f41710157.jpg)
![[ 34855-40-8] N-甲基-N-(丙烷-2-基)甲酰胺](https://www.cato-chem.com/files/2,2b92b571564b22.jpg)
![[ 128208-24-2] N-异丙基-N-甲基甲酰胺](https://www.cato-chem.com/files/4,2b92af4cc6126d.jpg)
![[1107650-56-5] 盐酸吗啉-d8](https://www.cato-chem.com/files/3,28a483eaf281ba.jpg)
![[19798-77-7] 4-氨基-3-氯吡啶](https://www.cato-chem.com/files/1,289e054f6fea93.jpg)
